MIT scientists have developed a machine learning model that proposes new molecules for the drug discovery process while ensuring the molecules it suggests can actually be synthesized in a laboratory.
Copyright: scitechdaily.com – “A Smarter Way To Develop New Drugs Using Artificial Intelligence”
 Pharmaceutical companies are using artificial intelligence to streamline the process of discovering new medicines. Machine-learning models can propose new molecules that have specific properties which could fight certain diseases, accomplishing in minutes what might take humans months to achieve manually.
Pharmaceutical companies are using artificial intelligence to streamline the process of discovering new medicines. Machine-learning models can propose new molecules that have specific properties which could fight certain diseases, accomplishing in minutes what might take humans months to achieve manually.
But there’s a major hurdle that holds these systems back: The models frequently suggest new molecular structures that are difficult or impossible to produce in a laboratory. If a chemist is unable to actually make the molecule, its disease-fighting properties can’t be tested.
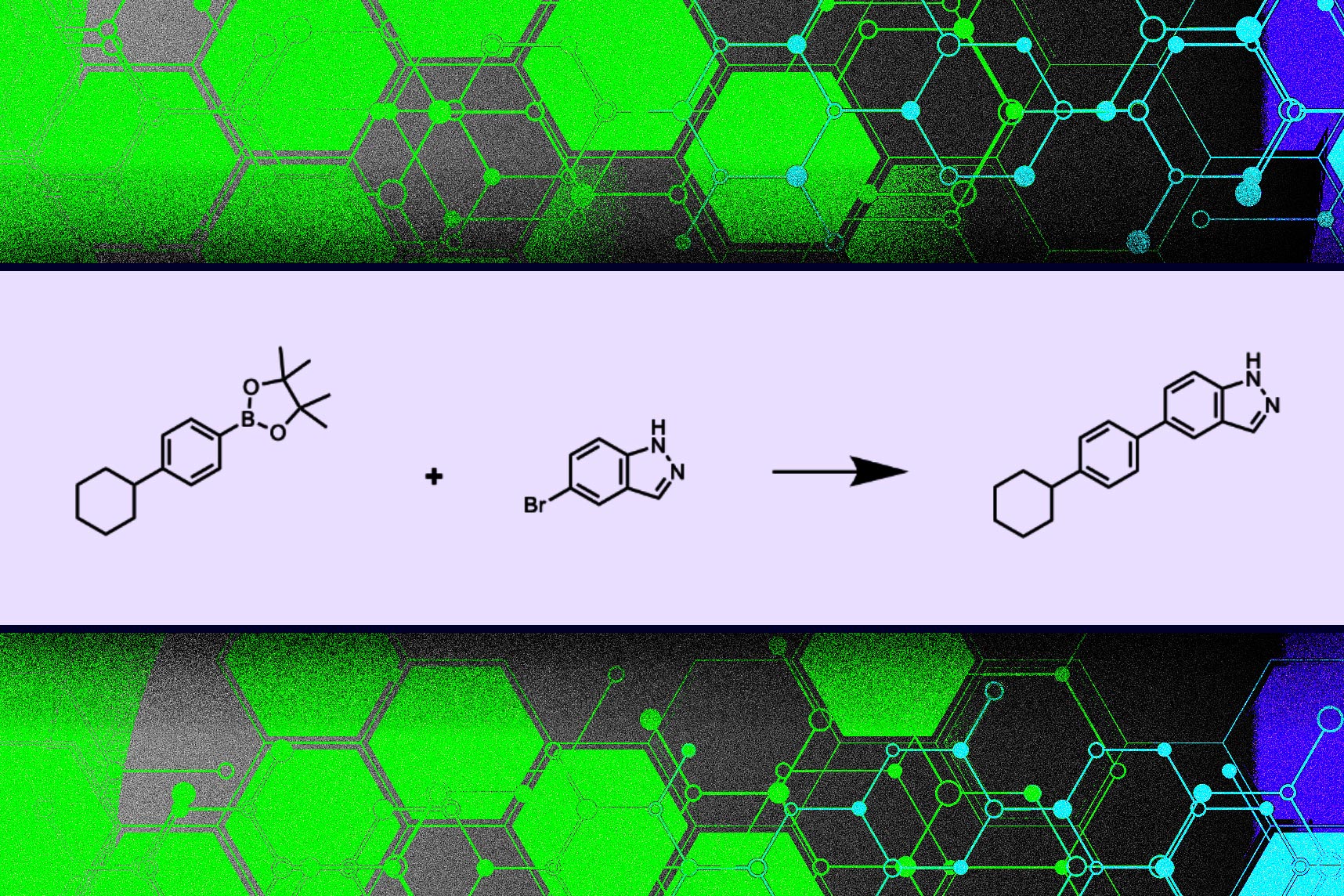
A new approach from MIT researchers constrains a machine-learning model so it only suggests molecular structures that can be synthesized. The method guarantees that molecules are composed of materials that can be purchased and that the chemical reactions that occur between those materials follow the laws of chemistry.
When compared to other methods, their model proposed molecular structures that scored as high, if not higher, on popular evaluations while also being guaranteed to be synthesizable. Their system also takes less than one second to propose a synthetic pathway, while other methods that separately propose molecules and then evaluate their synthesizability can take several minutes. Those time savings add up in a search space with billions of potential molecules.
“This process reformulates how we ask these models to generate new molecular structures. Many of these models think about building new molecular structures atom by atom or bond by bond. Instead, we are building new molecules building block by building block and reaction by reaction,” says Connor Coley, the Henri Slezynger Career Development Assistant Professor in the MIT departments of Chemical Engineering and Electrical Engineering and Computer Science, and senior author of the paper.
Thank you for reading this post, don't forget to subscribe to our AI NAVIGATOR!
Joining Coley on the paper are first author Wenhao Gao, a graduate student, and Rocío Mercado, a postdoc. The research was presented recently at the International Conference on Learning Representations.[…]
Read more: www.scitechdaily.com









MIT scientists have developed a machine learning model that proposes new molecules for the drug discovery process while ensuring the molecules it suggests can actually be synthesized in a laboratory.
Copyright: scitechdaily.com – “A Smarter Way To Develop New Drugs Using Artificial Intelligence”
But there’s a major hurdle that holds these systems back: The models frequently suggest new molecular structures that are difficult or impossible to produce in a laboratory. If a chemist is unable to actually make the molecule, its disease-fighting properties can’t be tested.
A new approach from MIT researchers constrains a machine-learning model so it only suggests molecular structures that can be synthesized. The method guarantees that molecules are composed of materials that can be purchased and that the chemical reactions that occur between those materials follow the laws of chemistry.
When compared to other methods, their model proposed molecular structures that scored as high, if not higher, on popular evaluations while also being guaranteed to be synthesizable. Their system also takes less than one second to propose a synthetic pathway, while other methods that separately propose molecules and then evaluate their synthesizability can take several minutes. Those time savings add up in a search space with billions of potential molecules.
“This process reformulates how we ask these models to generate new molecular structures. Many of these models think about building new molecular structures atom by atom or bond by bond. Instead, we are building new molecules building block by building block and reaction by reaction,” says Connor Coley, the Henri Slezynger Career Development Assistant Professor in the MIT departments of Chemical Engineering and Electrical Engineering and Computer Science, and senior author of the paper.
Thank you for reading this post, don't forget to subscribe to our AI NAVIGATOR!
Joining Coley on the paper are first author Wenhao Gao, a graduate student, and Rocío Mercado, a postdoc. The research was presented recently at the International Conference on Learning Representations.[…]
Read more: www.scitechdaily.com
Share this: